Intro
- Purification of DNA from sample
- Using combination of physical and chemical methods
- First DNA isolation done in 1869 by Frierich Miescher
- Routine procedure in molecular biology / forensice anlysis
- Chemical Method
- different kits used for extraction
- PCR sensitivity detection
- shows variation between commercial kits
Pocedure
3 Basic + 2 optional steps:
- Collect cells
- Break cell membranes a) Break lipids from cell membrane and nucleus with detergents and surfactants b) Break proteins with protease [optional] c) Break RNA with RNase [optionional]
- Treat solution with saline to make debris (broken proteins, lipids, and RNA) clump together
- Centrifuge to seperate clumped cellular debris from DNA
- DNA purification from detergents, proteins, salts and reagenze of cell lysis
- Ethanol precipitation
- Phenol-cholorofom extraction
- Mincolumn purification
Remove cellular and histone proteins bound to DNA either by:
- adding protease
- precipitate proteins with sodium or ammonium acetate
- extract with phenol-choloroform mixture prior to DNA precipitation
After isolation, disolve DNA in slightly alkaline buffer (usually TE buffer or ultra-pure water)
Nucleic Acid Preparation
Genomic DNA Preparation
DNA or RNA preparation is a prerequisite for the cloning of gene sequences as they exist in the genome and their transcripts, which may be processed.
Nucleic acid preparation is divided into several steps:
- Size reduction of the starting material and cell disruption.
- If necessary nucleus (organelles) preparation.
- DNA isolation from the digestion of the nuclear preparation or NA isolation from the total digestion according to one of the standard methods.
For the isolation of DNA as well as RNA four different approaches are used:
- Precipitation of NS from cell disruption
- Separation of a specific fraction with the help of CsCl gradient / density stage
- Ion exchanger columns
- Binding to silica gel [Melzak et al. (1996) Driving forces for DNA Adsorption to Silica in Perchlorate Solutions J. Colloid Interface Sci 181, 635-44]
For the lysis of the lipid bilayer of the cell membrane, a soap such as SDS is generally added to the buffer. If no DNA preparation from organelles (e.g. nucleus / mitochondria) is required, a chaotropic salt such as Guanidinium hydrochloride (see below) is usually used for the DNA preparation and RNA preparation. Organelle-specific preparations initially require an enrichment of this cell fraction.
Genomic DNA is required to isolate and analyze sections of the genome. The DNA must be highly molecular, i.e. it must not be broken by shear forces or degraded by DNase contamination. Tissues e.g. from liver, insects (Drosophila) or plants must be carefully digested beforehand, while cell culture cells are often taken up directly in buffers: For tissue digestion, the material cooled in liquid N2 is ground to a fine powder. The material is then further digested in a "Potter" vessel with buffer. If nuclear DNA is to be prepared without contamination with vesicle DNA (e.g. from mitochondria), this digestion is performed in an isotonic SDS / Tris-Cl buffer. Then, after separation of undissolved components, the digestion is applied to a higher density solution, e.g. a 2M "sucrose step". The heavy nuclei are pelletized by centrifugation, the rest remains above the sucrose stage.
For the preparation of unshredded, high-molecular nuclear DNA, the protein structure is dissolved at elevated temperature in buffer / SDS (50o-65oC) with the addition of proteinase K and the DNA is released. This avoids shear forces and proteins including DNases are degraded by Proteinase K. After protein / SDS removal by phenol / chloroform extraction, the DNA is precipitated by addition of alcohol in the presence of monovalent cations, preferably NaOAc. 0.5 - 1.0 volume (vol.) isopropanol precipitates high-molecular DNA (including RNA) in contrast to 2.0 - 2.5 volume ethanol without salt co-precipitation. Afterwards it must be "washed" with 70% ethanol to remove residues of isopropanol as it can inhibit enzymes.
If special nuclear DNA is not required, the digestion is carried out in high concentration of a chaotropic salt such as guanidinium isothiocyanate (GITC), which leads to the denaturation and dissolution of proteins and thus immediately inactivates DNases and RNases. DNA and RNA can be isolated from this digestion using one of the above methods.
What are chaotropic salts (Macherey-Nagel, BioNews 7): The term goes back to the Hofmeister series (F. Hofmeister (1888) Zur Lehre von der Wirkung der Salze, Arch. Exp. Pathol. Pharmacol. (Leipzig) 24, 247-260), which describes the ability of ions to destroy the structure of proteins. Chaotropic ions are large single-charged ions with low charge density. All show a weaker interaction with water molecules than the interaction of the water molecules with each other. Instead, they increasingly attach themselves to macromolecules such as proteins. This makes the network of hydrogen bridge formation into macromolecules that interact with the chaotropic salt and dissolve its structure.

Total DNA of high purity is obtained by "banding" the digested material in approx. 5M CsCl Lsg. in an isopycnic gradient over >16h instead of isopropanol precipitation. The salt solution forms a density gradient in which protein with a lower density accumulates at the top, RNA with a higher specific density than DNA appears at the bottom and DNA is concentrated in the middle as a white band. This procedure is costly and time-consuming, but results in particularly clean preparations.
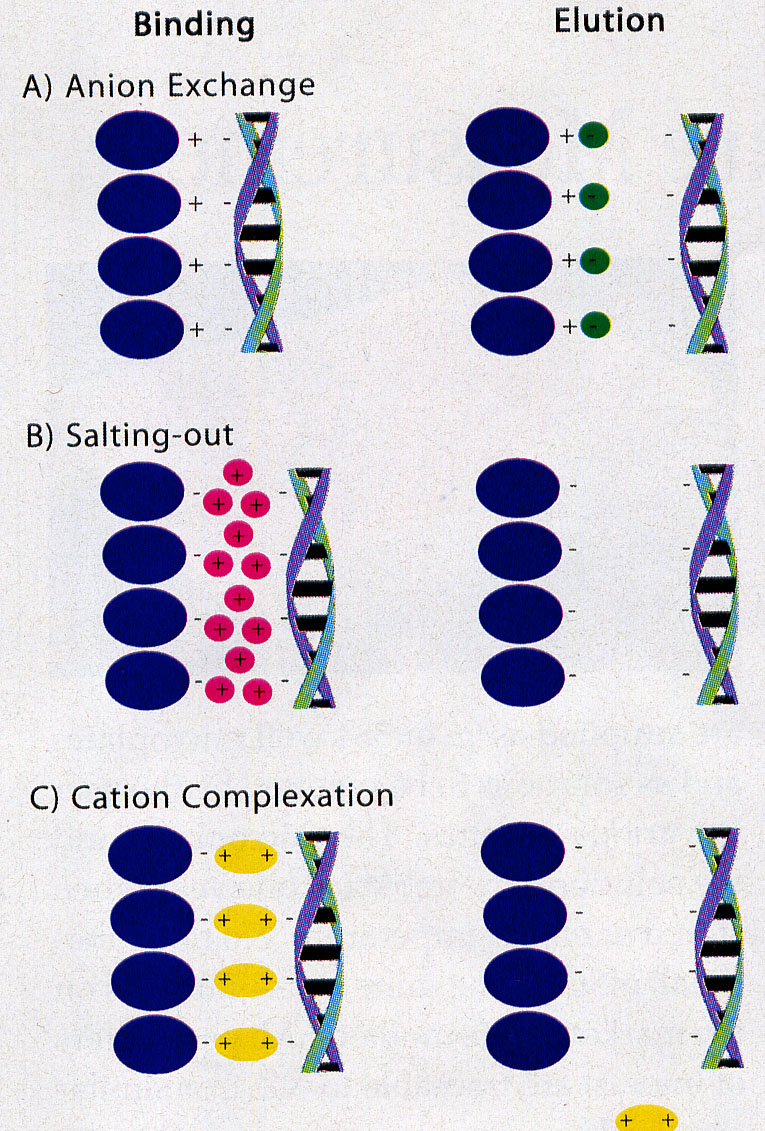
Comparable results can be obtained by using ion exchanger columns for DNA preparation from the digestion. As a rule, exchangers containing diethylaminoethyl or methylaminoethanol pendant groups - usually on a silicate basis - are used. Very small volumes must be maintained in order to work on a micro scale. Therefore, a high binding capacity of the exchange material is required, which is achieved by dense loading with reactive exchange groups together with an extremely small particle size. To bind the DNA, the salt concentration must be well below the concentration that displaces the binding at the ion exchanger. This value is about 1.5M KCl for double-stranded DNA. However, this concentration is so high that the DNA salt can be precipitated with the subsequent alcohol precipitation. Therefore 15% ethanol is added to all solutions. The addition of 15% ethanol (= approx. 3M) in all buffers has the effect that the "hydrate shells" for the solubility of the K+ / Cl ions are reduced. This leads to an "effective" salt concentration increase. This allows you to work with significantly lower salt concentrations with the same effect.
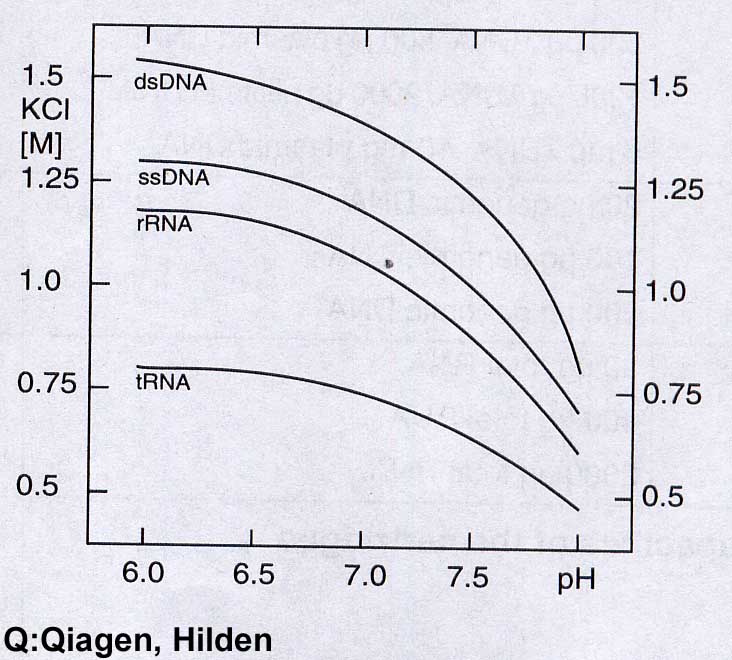
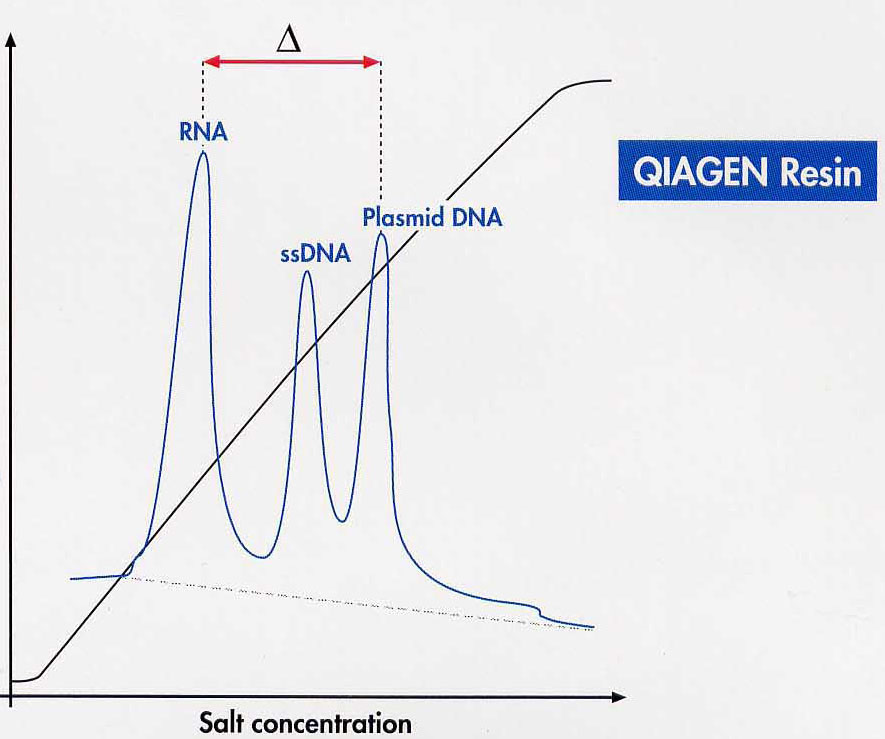
In concrete terms, this means that 0.8 to 0.9M salt concentration / 15% ethanol is used to disrupt the cell to bind the DNA to the column. You can wash all NS except dsDNA from the column with up to approx. 1.1M KCl / 15% ethanol, pH 6.3. By applying a salt gradient the different NA can be desorbed separately. However, the fractional desorption of the different NS from the ion exchanger is hardly carried out.
Nucleic acids bind better at pH 6.3 than at pH 8.5, since the ammonium group of the exchanger is significantly more positively charged at pH 6.3 than at pH 8.5. Therefore, lower salt concentrations can be used for DNA displacement at pH 8.3. DsDNA is already released from the column by 1M KCl / 15% ethanol, pH 8.3 after a pH change.
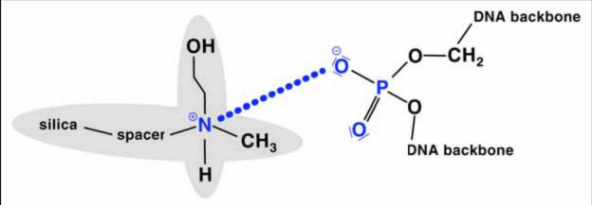
If only a few g quantities of genomic DNA are required, it makes sense to use a "spin column" rapid method. NA, i.e. DNA and RNA, are selectively bound to a matrix by GITC after digestion of the tissue. RNA can be removed by adding DNase-free RNase directly to the column. Two materials are in use: clay and silica gel. Both materials bind NS at high ionic strength. High ionic strengths are automatically present when the cell material has been digested in a chaotropic salt.
Spin columns containing silica gel (Qiagen, Roche) are the most widespread. After washing with 80% ethanol, NS with TE (10mM Tris / Cl, pH 7.8, 1mM EDTA) or H2O are eluted by centrifugation into an Eppendorf tube.
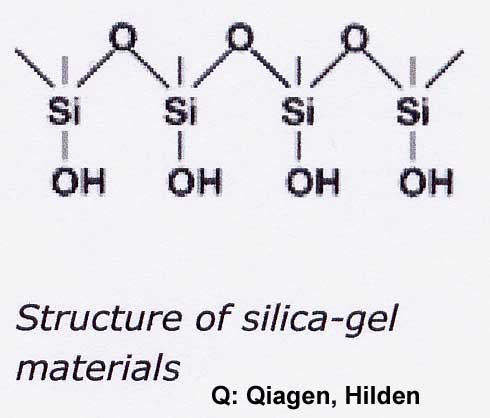
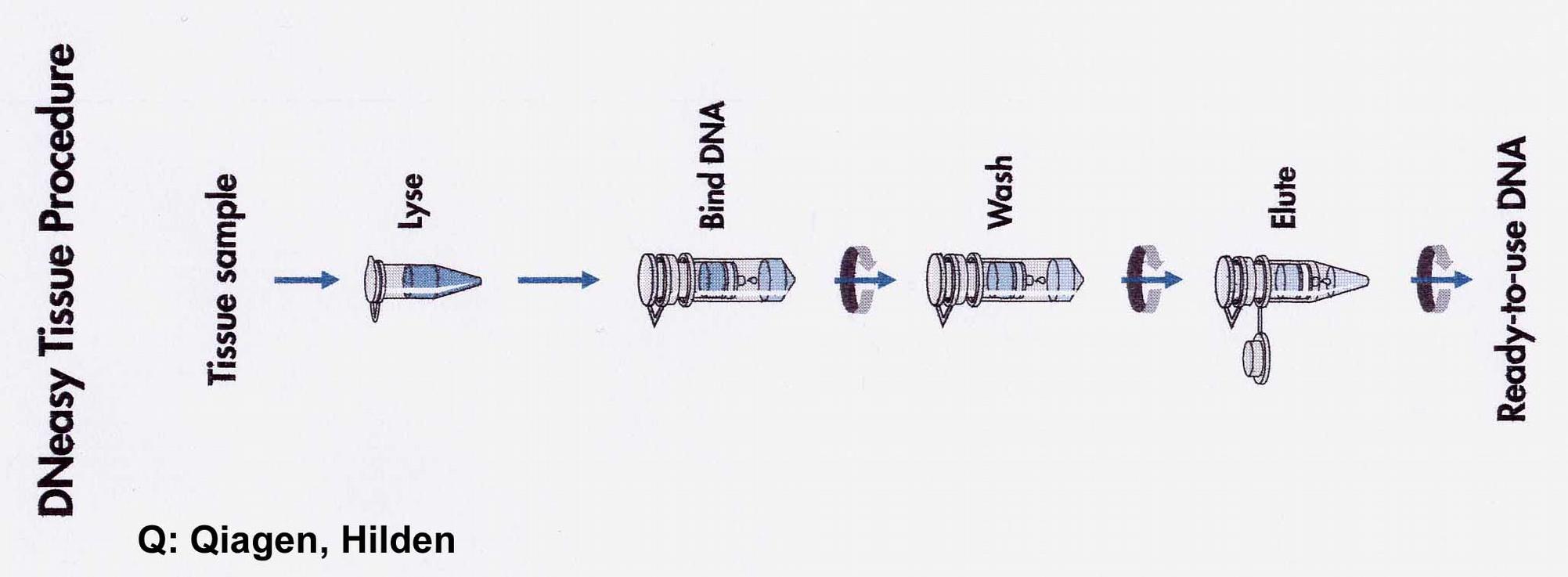
Two theories for the binding of NA to silica gel are discussed (Macherey-Nagel, Bio News 7). On the one hand, the formation of hydrogen bonds between DNA and silicate is required after exclusion of the hydrate shell. On the other hand, it is believed that nucleic acids adsorb to the silica gel by a salt-out effect as long as water is removed from the dipole by high salt concentration and ethanol. The Guanidinium ion saturates the charges between silicate and NS. In the washing step, the high salt concentration is replaced by 80% ethanol (16M compared to approx. 11M H2O in the solution - water removal prevents solution!). Polysaccharides and proteins do not bind and can be washed out. Even single-stranded NS under a length of 100 nucleotides (oligonucleotides, small RNAs) are not bound.
A new development are the Spin-Columns containing clay / sand. NS bind under high salt in the presence of 50 - 100mM Mg2+ by complex formation between the negatively charged carrier material and the negatively charged backbone of NS. After a washing step with an alcohol-containing buffer the NS are released with TE.

Compared to the preparation of genomic DNA, the preparation of plasmids or phages requires different strategies because the DNA has to be isolated from the bacterial chromosome. Direct precipitation of plasmid DNA from cell disruption separated from genomic DNA is not possible. Phages are usually released into the medium containing DNA of lysed bacteria. Therefore, methods have been optimized to separate the contaminating bacterial DNA as early as possible. The process must continue to be "simple" enough to be suitable for high throughput.
Plasmid DNA Preparation
For the investigation of cloned DNA a mini-preparation from 1.5ml culture volume is usually sufficient, which should yield approx. 10-20g DNA. In rare cases a midi preparation of 50ml culture volume with a yield of 100 - 200g DNA is necessary (see below). The digestion method in alkaline pH has established itself: Starting from 1.5ml overnight culture for mini preps, the pellet from the overnight culture, OD600 > 2.0, is resuspended in Tris / Cl buffer. In part, lysozyme is added to the E. coli cell wall to cleave the murein network, as in the original regulation [H.C. Birnboim und J. Doly (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA, Nucl. Acids Res. 7, 1513-1523.].
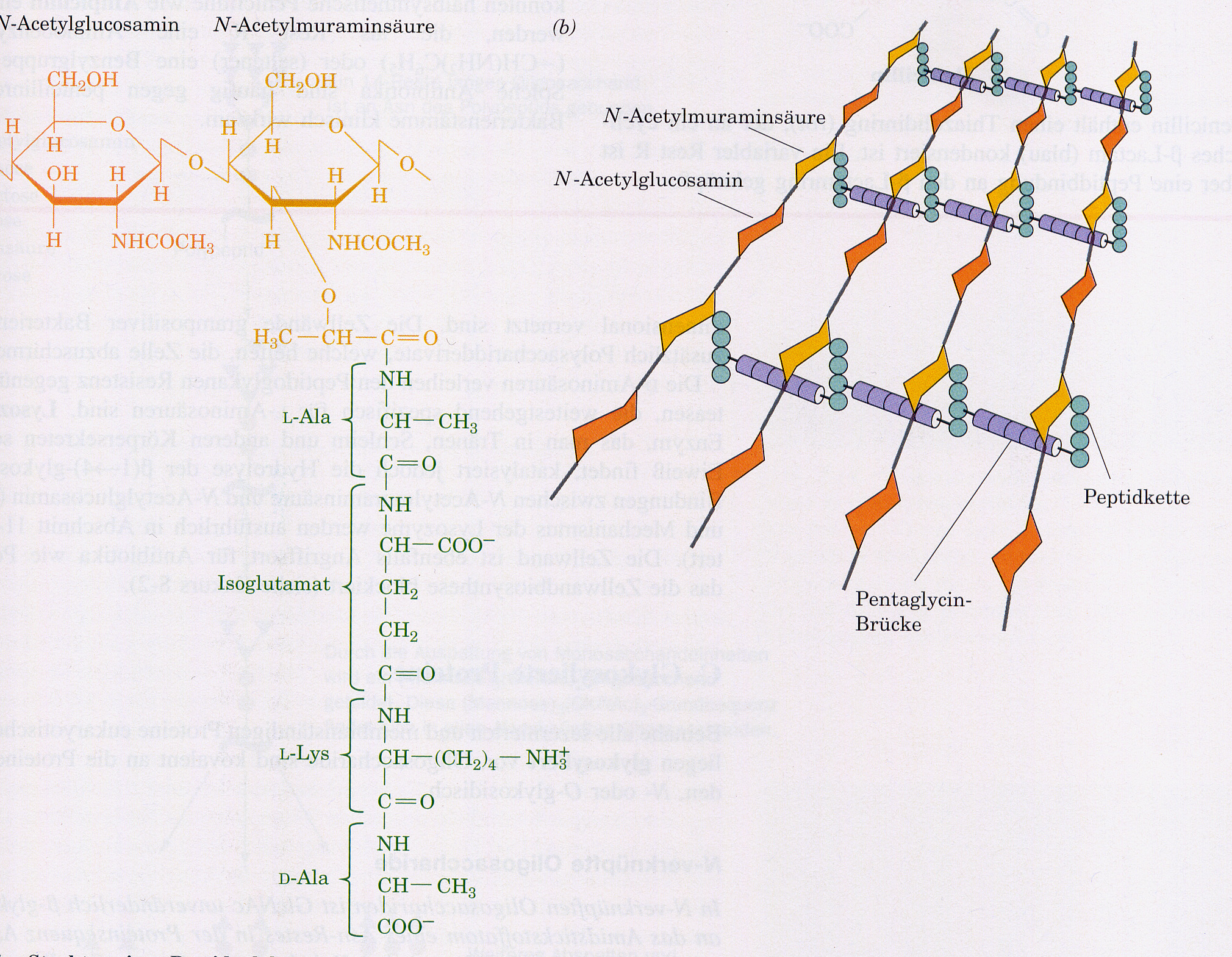
It hydrolyses the glycosidic (1-4) bond between N-acetylmuraminic acid. Then it is lysed with 0.2M NaOH/1% SDS. SDS/NaOH dissolve the lipid bilayer of the cell membranes and denature the E.coli "chromosome" and the protein. OH- and addition of RNaseA largely degrade the RNA components. Then K-acetate, pH5.1, is neutralized with 3M and the SDS is precipitated as a poorly soluble potassium salt. The bacterial chromosome, which is anchored to the cell membrane with its origin of replication, can at best partially renature and forms an insoluble mass with the membrane and protein components. According to Birnboim and Doty, plasmid DNA is in "covalently closed circle" form, at approx. pH 12.5 in contrast to high-molecular E.coli DNA not yet denatured. It is more likely that a plasmid ring in CCC form will easily renaturate due to spatial proximity. After centrifugation of the insoluble components, the plasmid is therefore present in the supernatant. In the classical mini-prep process, the plasmid DNA is precipitated directly with 0.6 vol. isopropanol. The pellet is washed with 70% ethanol and then absorbed into TE buffer. Available mini-preparation kits improve the purity of the DNA with little effort, e.g.: "QIAprep" (Qiagen) The supernatant of the K-acetate precipitation (0.9M K-acetate) is added to a spin column with a silica gel membrane. With low salt buffer the total nucleic acids are obtained.
"Cation Complexation Technology" (CCT, Molzym) The supernatant of K-acetate precipitation is applied to a CCT Spin Column Technology (PrestoSpin D) in the presence of 50-100mM Mg2. After washing with isopropanol and ethanol, the plasmid DNA is desorbed with TE.
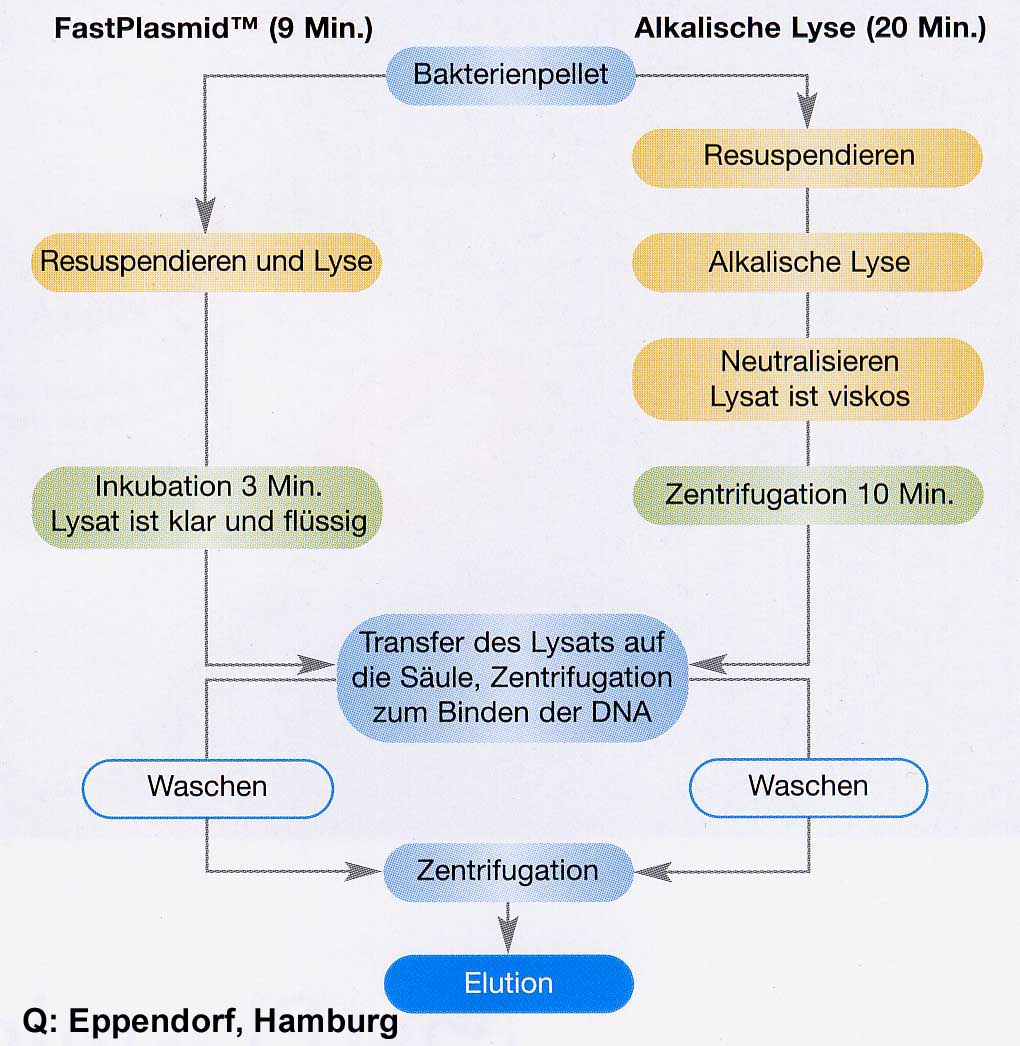
Alternative method: "FastPlasmid" (Eppendorf) introduces another digestion method without NaOH / K-acetate in combination with a coarser-meshed silica gel membrane. E.coli cells are taken up with a buffer containing salt / RNaseA / lysozyme. The lysozyme activity destroys the murein network only slightly, so that holes are formed in the cell wall. High external salt concentration leads to an osmotically induced "emptying" (bursting of the cells) of the cell contents including plasmid DNA. RNaseA destroys RNA contamination. The lysate is applied directly to a spin column and washed with 55% isopropanol. All non-binding components including the plasmid-depleted E.coli cells with a size of 1-2m are washed / centrifuged through the membrane with the bacterial chromosome, since the pore size of this special membrane is approx. 4m. With 80%ethanol residues of isopropanol are washed out and with 10mM Tris / Cl, pH 8.0, the plasmid DNA is desorbed free of genomic DNA.
For the preparation of plasmid DNA in mid-scale (<100ml culture), the supernatant of the K-acetate precipitation (see above) is precipitated onto larger ion exchange columns and the preparation of genomic DNA is performed as in VL02. Alternatively, plasmid DNA is separated from residues of chromosomal bacterial DNA by an isopycnic CsCl gradient. However, this requires a process that gives the two types of DNA different densities. This is achieved by their different binding capacity of ethidium bromide (EtBr), which is added in high concentrations. EtBr moves between the base stacks. This stretches the DNA. The more EtBr is bound, the lighter the DNA becomes. Plasmids in E.coli are "undermined" by topoisomerases. But since "B-DNA" is If a helix turn / 10.4 nucleotides is attempted, either a partial unwinding or, since it is more energy-efficient, a negative super spiralization occurs.
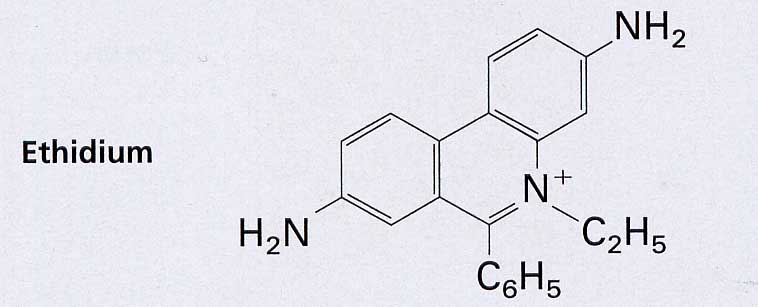
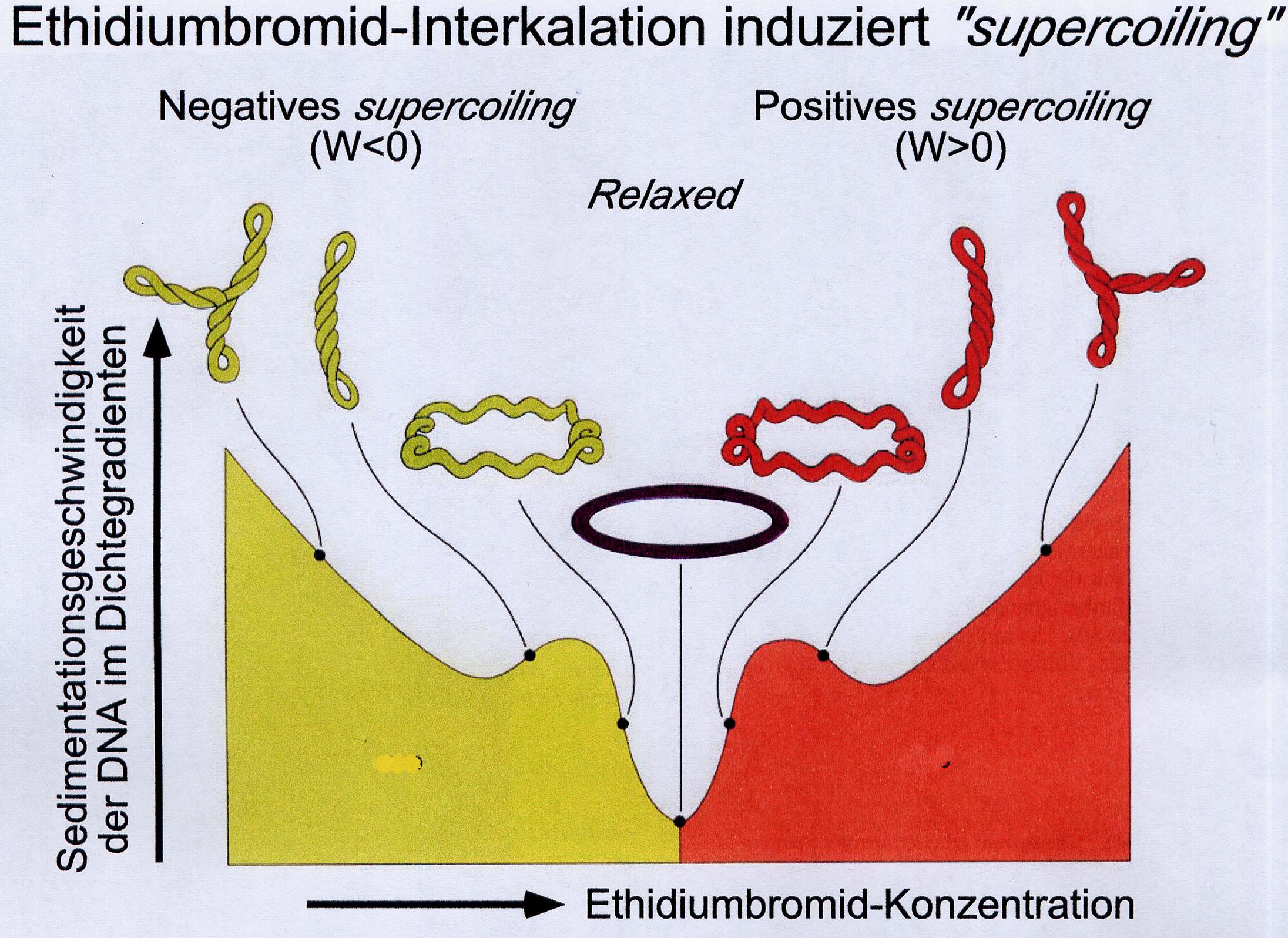
EtBr storage reduces the helical winding and thus eliminates the negative superspiralization and finally converts it into a positive one. The storage of further EtBr is limited by the only limited possible positive superspiralization in the closed ring system. Chromosomal bacteria THEN, as far as they are present at all after alkaline digestion, can store far more EtBr, since they contain nicks due to their size and are therefore no longer subject to any corresponding topological tension. With increasing EtBr storage, its specific density becomes lower than that of plasmid DNA. In the density gradient, it forms a band in the "higher", i.e. lighter, range. The plasmid DNA can be withdrawn with a syringe and precipitated with isopropanol after extraction of the EtBr with butanol.
λ Phage DNA Preparation
Lytic growth of phages means that E.coli cells are lysed and no cells have to be disintegrated to isolate the phages. Phage DNA can be easily prepared by isolating phage particles. This can be done in two ways: the production of fully lysed bacterial turf ("plate lysate") or a liquid culture of E.coli, which should only be lysed at highest bacterial density.
The lysate still contains impurities at cell debris (with E.coli DNA and RNA) and unlysed cells, which are first centrifuged. To remove NA residues from lysed E.coli, the phage suspension is incubated with DNase / RNase. The phage particles are then precipitated. This is achieved by adding a polyether, such as polyethylene glycol (PEG), which binds enough water molecules to remove the solubility of the phage particles. The phage is pelletized and ready for DNA processing. The phage shell is broken open by adding detergents (SDS) at 65oC and the phage DNA is released. It can be isolated with anione exchangers columns (see above) or precipitated directly.
RNA-Preparation
"Mature" RNA transcripts in eukaryotes are often not colinear with the gene sequence. Therefore, the sequence of the RNA is of great importance for the molecular genetic investigation of gene expression. Only then is it possible to determine which exons have been combined with each other. For cloning, the RNA has to be transcribed into a DNA copy (cDNA). This requires the preparation of intact RNA. In order to isolate intact RNA from tissue, the activity of internal RNases must be excluded during tissue disruption. This is only possible by denaturing the proteins during cell disruption (tissue, animals) and by strictly observing precautionary measures when working with RNA (see below). Cell disruption is performed in the presence of Guanidinium isothiocyanate. RNase-free water should be used as soon as the RNA preparation is included in Guanidinium-free buffer solutions. Addition of 0.1% diethylpyrocarbonate (DEPC) overnight inactivates RNases. Residues must be inactivated by heating - decomposition into CO2 and ethanol.
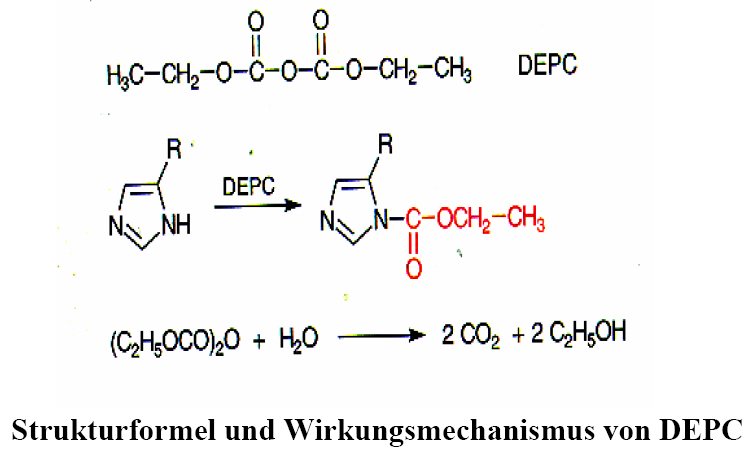
DEPC acts on RNases that contaminate buffers or vessels by reacting with a histidine of the active centre to N-carbethoxyhistidine; however, it also reacts with nucleic acids, so it must have been completely removed by heating before using the treated water.
For the isolation of RNA after GITC digestion either a density gradient centrifugation (CsCl step) or RNA (especially >100Nt) can be selectively precipitated from DNA-containing solutions by 2.5M LiCl / 0oC. The Internet page: http://www.ambion.com/techlib/tb/tb_160.html shows that also lower LiCl-concentrations are sufficient. For the density gradient method, the clear supernatant of the cell disruption is given to a "CsCl level" higher than the density of DNA and centrifuged overnight. Only RNA pellets to the soil because it has a higher specific density than the CsCl level. After removing the supernatant, it can be taken up in water. This method leads to RNA of excellent quality, but is too time-consuming and too expensive for routine operation.

The method of choice is acid phenol extraction ["TRIzol-Methode", Chomczynski and Sacchi (1987) Anal. Biochemistry 162, 156-159]. Triozole is a single-phase mixture of GITC and phenol in a weakly acidic buffer (pH 4.0). In this method, the digestion of the mortarized cell material TRIzol is directly converted into a separation of protein and DNA. Phenol is soluble in a highly molar aqueous GITC solution without phase separation. Only the addition of chloroform leads to phase separation of the TRIzol solution. Thus, the proteins unfolded by GITC are transferred into the organic phase. At the slightly acidic pH of the buffer (approx. 4.0), DNA also passes into the phenol phase. RNA remains specific in the aqueous phase and can be precipitated with isopropanol. From the phenol phase the DNA can be precipitated with ethanol; the protein can still be precipitated from the phenol / ethanol supernatant with isopropanol [Hummon et al., (2007) Biotechniques 42, 467-472].
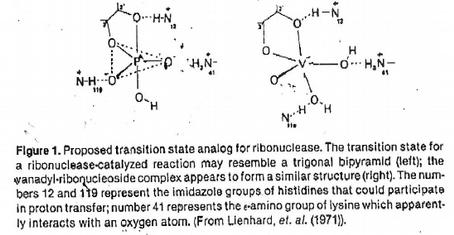
The RNA RNase is endangered after the uptake of the RNA pellet in buffer. One can RNase I have to admit to inhibitors. The oxovanadinium (IV) ion (VO2+) in its hydrated form forms complexes with each of the four ribonucleotides (see above) in the form of a trigonal bipyramid. Thus they represent analogues of the 2',3' cyclic transition state of RNA hydrolysis and bind firmly to the active centre of the RNases [(G. Lienhard et al. (1971) Cold Spring Harbor Symp. Quant. Biol. 36, 45-51].
To isolate small amounts of RNA, GITC digestion can be applied directly to a silica membrane spin column, which results in a selective binding of RNA >200NT. The separation of chromosomal DNA is possible with RNase-free DNase directly on the membrane.
Poly(A+)RNA Preparation
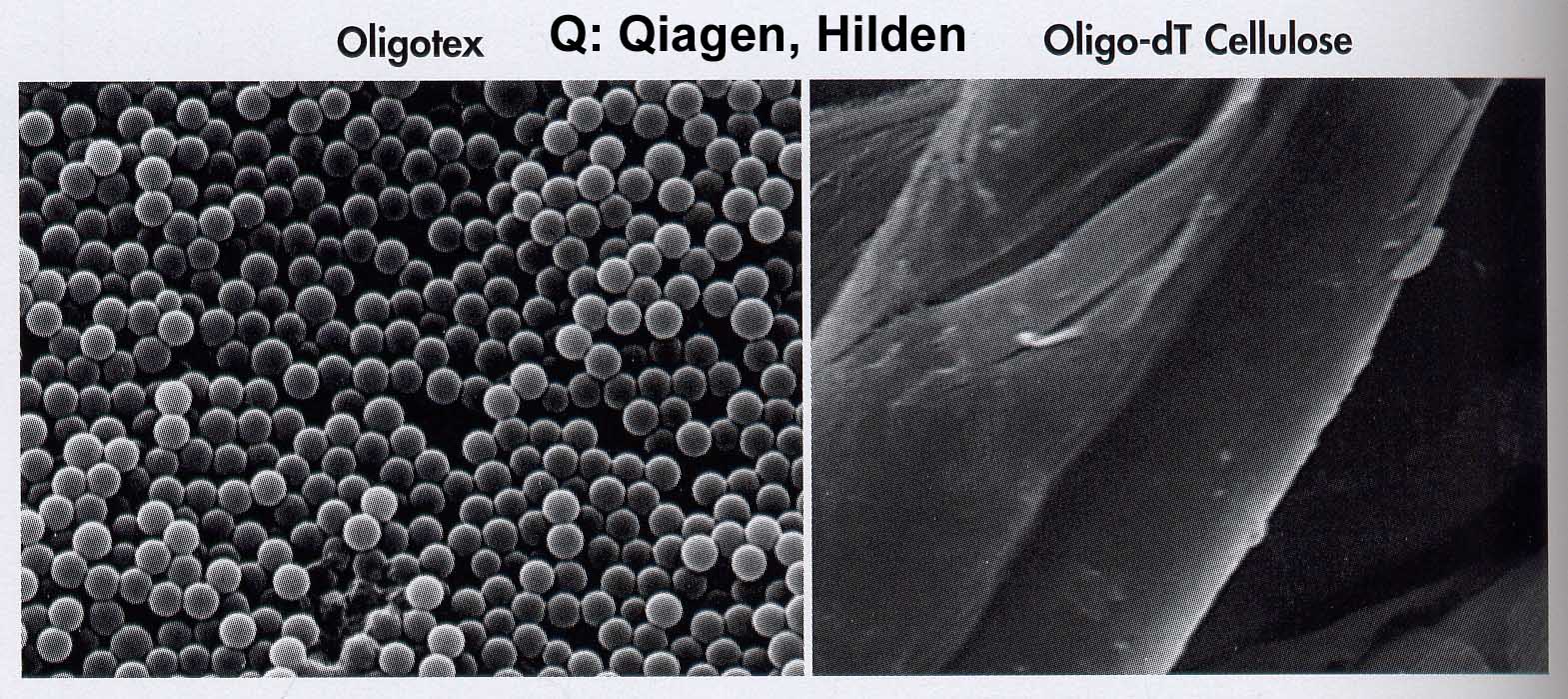
The polyadenylated mRNA population is isolated affinity chromatographically from the total RNA. Oligo-dT cellulose or poly-U sepharose are the classical matrices for the binding of poly(A+)-RNA. The binding capacity has been dramatically increased by the development of a new carrier material.
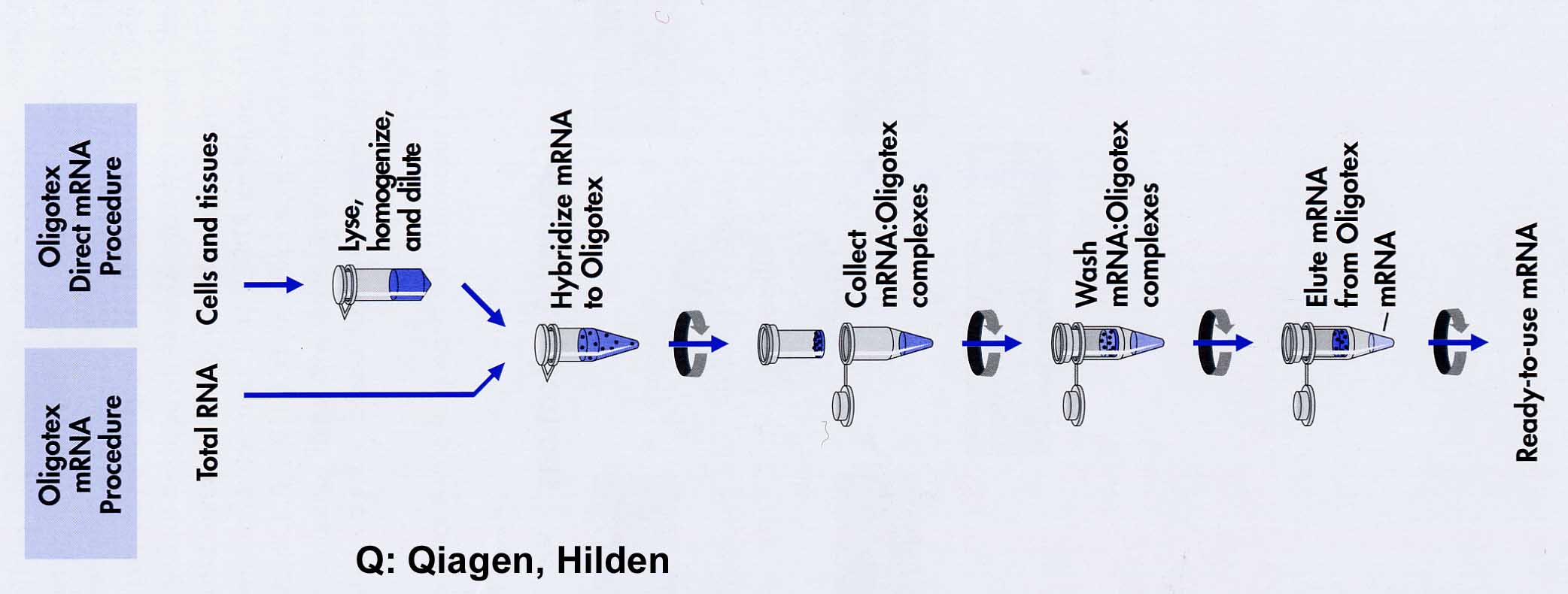
Oligotex consists of 1.1m polystyrene latex beads which have been chemically coated with dC10T30. This allows an extremely large surface and oligodT density / volume. Therefore it is possible to work with this substance in small volumes in the Eppendorf vessel (2 powers of ten volume reduction): The mRNA is bound in 0.5M NaCl with its poly(A+)-tail to oligodT by hybrid formation. After centrifugation, the supernatant is removed and unspecific bonds are washed out at an intermediate salt concentration of 150mM NaCl. With H20 or 5mM buffer the poly(A)-RNA is dissolved at 70oC. A very successful alternative is the use of magnetic particles. Oligo-dT particles are provided with magnets so that the washing step is reduced to fishing out the particles with magnets attached at the side.
Questions
- Under which salt conditions (high or low) do nucleic acids bind to ion exchangers (1) and silica membranes (1)?
- How can you completely remove primers from a PCR approach, two ways (2)?
- With an acidified phenol buffer solution they can isolate RNA from a cell disruption. Where are protein and DNA (Explanation, 4)?
- Under which salt conditions do you bind your poly (A)+RNA fraction to an Oligotex column (1)?
- How to prepare lambda phage on agar plates (three steps, 3)?
- Why are the binding strengths of nucleic acids at DEAE ion exchange columns pH dependent (2)?
- Name the three crucial steps of a mini preparation of DNA (3 x 1) and describe the effect of each step (3 x 1).
- How does the specific weight of DNA change when EtBr (2) is stored?
- Which bond of the murein network of E.coli cleaves lysozyme (2)?
- How do the salt conditions for binding and detaching nucleic acids on ion exchangers differ from those of silica gel (4)?
- Why are chaotropic saline solutions such as GITC preferred for cell disruption in nucleic acid preparation (explanation 3)?
- Which nucleic acid types (RNA, DNA) of which size are bound by silica spin columns (1)?
- Where can you find RNA, DNA and protein in an isopycnic CsCl gradient (3)?
- What effect does the addition of alcohol (e.g. 15% ethanol) have on the "active salt concentration" of a solution (2)?
- Why can E.coli DNA be separated from plasmid DNA by alkaline digestion using the "Miniprep" method (explanation, 2)?
- Which nucleic acids are bound by silica gel spin columns (2)?
- What effect is the FAST Plasmid Miniprep process (1) based on?
- In the chemical synthesis of ribo-oligonucleotides, the problem arises that the protective groups of the primary amino groups of the bases have to be cleaved off in the alkaline pH. Why is this possible even though RNA is alkalilabile (explanation, 4)?
- With an acidified phenol buffer solution they can isolate RNA from a cell disruption. Where are protein and DNA (Explanation, 4)?
- You have amplified a DNA fragment with PCR; you have present this fragment in the PCR reaction solution and want to amplify a section with a new primer pair. How do you proceed in order to achieve this goal as quickly and as easily as possible (2)?
- Trizole combines properties that make it particularly suitable for the isolation of RNA. What are these two components and how can ungraded RNA be obtained directly from protein and DNA (Explanation, 4)?
- Which vanadyl salt complexes are used as RNase inhibitors (1), what does the binding to RNase look like (2)?
- Under which salt conditions would they bind their total RNA to an Oligotex column for poly(A)+ selection, under which conditions (salt concentration/temperature) would they elute the RNA from the column (3)?
- Name the three steps of the TRizol RNA preparation procedure (3).
Material
- Test tubes
- Falcon Tubes
- Eppendorf Tubes
- Pipette
- Centrifuge
- Vortex
Flair: Science
Collection: Biology Crash Course
